1. 复旦大学生命科学学院 上海 200438; 2. 上海市公安局青浦分局刑事科学技术研究所 上海 201700;
摘要 目的:探究基因组DNA污染及乳腺癌单样本预测器(single sample predictor, SSP)在RNA-seq中鉴定Basal-like亚型乳腺癌的一致性与稳健性。方法:使用4种SSP联合3种数据校正方法,按比例随机去掉一定比例的非Basal-like亚型样本,并使用正态分布模拟基因组DNA对基因表达量的影响。结果:数据校正方法影响乳腺癌SSP鉴定Basal-like亚型的一致性(κ=0.733-0.964);乳腺癌SSP可以一致鉴定出Basal-like亚型(κ=0.846-0.958),对Basal-like亚型鉴定的稳健性高;基因组DNA污染对Basal-like亚型鉴定的一致性影响较小(κ=0.922-1)。结论:Basal-like亚型的鉴定具有高度的一致性与稳健性且受基因组DNA污染影响较小。
关键词:基因组DNA污染;RNA-seq;乳腺癌分型;Basal-like亚型
背景
关于乳腺癌目前形成了存在5种分子亚型的共识:Basal-like, Her2-enriched, Luminal A, Luminal B及Normal-like。其中Basal-like亚型的乳腺癌具有发病年龄小,恶性程度高,预后较差,与其他乳腺癌亚型相比,Basal-like亚型缺少有效治疗方案。由于Basal-like亚型难治性的特点,如果对Basal-like与非Basal-like亚型在临床上加以区分,便可以为医生在对病人进行临床决策时提供参考。
用于乳腺癌分子亚型鉴定的乳腺癌单样本预测器(SSP)1-4可以在芯片(Microarray)数据中一致鉴定出Basal-like亚型乳腺癌5,乳腺癌分型数据集的组成则会影响样本的分子分型结果4, 6,而这些结论是否适用于RNA-seq数据仍需探究。此外,基因表达定量技术受到基因组DNA污染的影响7, 8,临床样本测序得到的RNA-seq数据更易受基因组DNA污染的影响,这些污染是否会影响Basal-like亚型的鉴定目前尚未有研究。在本研究中,我们使用The Cancer Genome Atlas (TCGA)数据库中的原发性乳腺癌,利用正态分布模拟基因组DNA污染及两例已知基因组DNA污染浓度的样本,来探究Basal-like亚型乳腺癌鉴定时的一致性与稳健性及基因组DNA污染对其鉴定的影响。
方法
数据来源
TCGA数据库乳腺癌项目中1093例原发性乳腺癌及2例已知基因组DNA污染浓度(0%,1%)的RNA-seq数据,表达量标准化方式分别为RSEM及FPKM。
数据校正方法
数据校正方法分别为:无校正,gene-centering 9(GC)校正及R包genefu10中的基于分位数校正。
乳腺癌分型SSP
基于基因表达量的平均值的SSP:SSP20031, SSP20062, PAM503及基于基因表达量大小关系的AIMS4。本研究中AIMS只使用未校正数据产生分型结果4。
改变乳腺癌分型数据集组成
先根据整个数据集分型结果将样本分为Basal-like及非Basal-like亚型(数据校正方法为GC),再按比例从数据集中随机去掉部分非Basal-like亚型样本。
模拟基因组DNA污染
使用正态分布N(0,1)对所有样本产生长度为总基因个数的向量,将这些向量与RSEM数据相加即得到模拟的受基因组DNA污染的表达谱。
统计分析
TCGA数据下载使用TCGA2STAT包中getTCGA()函数。Basal-like亚型一致性评估使用Cohen’s κ coefficient5,该值区间在0.81-0.99代表“almost perfect agreement”,使用base包中kappa()函数计算。GC校正使用base包中scale()函数。分位数校正使用genefu包中rescale()函数。分型使用genefu包中intrinsic.cluster.predict()函数及AIMS包中的applyAIMS()函数。正态分布模拟使用rnorm()函数。所有统计分析基于R 3.6.1。
结果
数据校正方法影响Basal-like亚型鉴定的一致性
使用3种数据校正方法对数据进行校正后的分型结果显示,不同数据校正方法均影响Basal-like亚型鉴定一致性(表1)。
表1. 不同数据校正方法影响Basal-like亚型鉴定的一致性
校正方法1 | 校正方法2 | κssp2003 | κssp2006 | κPAM50 |
未校正 | GC | 0.946 | 0.733 | 0.894 |
未校正 | 分位数 | 0.964 | 0.780 | 0.939 |
GC | 分位数 | 0.962 | 0.950 | 0.955 |
κ= Cohen’s κcoefficient.
不同SSP可以一致鉴定出Basal-like亚型
不同SSP间鉴定Basal-like亚型的一致性均达到“almost perfect agreement”(表2)。样本个体水平,不同的SSP可将同一样本高度一致地鉴定为Basal-like亚型(图1)。
表2. 不同分型方法之间均可以一致鉴定出Basal-like亚型
分型方法 | 分型方法 | 校正方法 | κ | 校正方法 | κ | 校正方法 | κ |
SSP2003 | SSP2006 | 未校正 | 0.870 | GC | 0.846 | 分位数 | 0.879 |
SSP2003 | PAM50 | 未校正 | 0.947 | GC | 0.889 | 分位数 | 0.937 |
SSP2003 | AIMS* | 未校正 | 0.958 | GC | 0.938 | 分位数 | 0.949 |
SSP2006 | PAM50 | 未校正 | 0.889 | GC | 0.885 | 分位数 | 0.911 |
SSP2006 | AIMS* | 未校正 | 0.855 | GC | 0.861 | 分位数 | 0.904 |
PAM50 | AIMS* | 未校正 | 0.951 | GC | 0.918 | 分位数 | 0.956 |
κ= Cohen’s κcoefficient.
*只使用未校正数据产生的AIMS分型结果
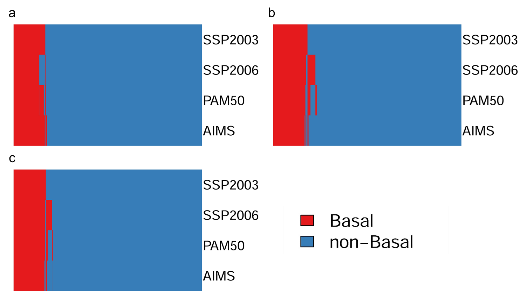
图1. 不同数据校正方法分型结果。a, b, c) 分别为未校正、GC校正及分位数校正数据的分型结果。行代表SSP,列代表样本;红色表示Basal-like亚型,蓝色代表非Basal-like亚型。
不同SSP对Basal-like亚型鉴定的稳健性高
随机去掉一定比例非Basal-like样本后的分型结果显示,不同SSP对Basal-like亚型鉴定的稳健性高(图2)。
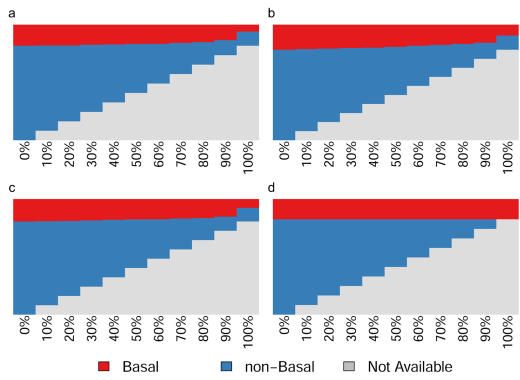
图2. 不同非Basal-like亚型比例下的分型结果。其中a, b, c, d) 分别表示SSP2003, SSP2006, PAM50及AIMS分型结果。行代表样本,列代表去掉的非Basal-like亚型样本比例;红色表示Basal-like亚型,蓝色表示非Basal-like亚型,灰色表示去掉的样本。
基因组DNA污染对Basal-like亚型鉴定影响较小
比较模拟基因组DNA污染前后的分型结果,发现基因组DNA污染对Basal-like亚型的鉴定影响较小(表3)。两例已知基因组DNA污染浓度的样本在AIMS方法中被鉴定为Basal-like亚型,在其他方法中均被一致鉴定为非Basal-like亚型。
表3. 基因组DNA污染对Basal-like亚型的鉴定影响较小
分型方法 | 校正方法 | κ | 校正方法 | κ | 校正方法 | κ |
SSP2003 | 未校正 | 0.985 | GC | 0.959 | 分位数 | 0.959 |
SSP2006 | 未校正 | 0.926 | GC | 0.922 | 分位数 | 0.941 |
PAM50 | 未校正 | 0.970 | GC | 0.938 | 分位数 | 0.967 |
AIMS | 未校正 | 1 | GC | 1 | 分位数 | 1 |
讨论
本研究探究了乳腺癌样本分型时,基因组DNA污染及不同数据校正方法及不同SSP对鉴定Basal-like亚型的一致性与稳健性的影响。结果显示,不同数据校正方法影响Basal-like亚型鉴定的一致性,但一致性仍较高(表1)。不同SSP对Basal-like亚型鉴定的一致性高(表2,图1)及稳健性高(图2),其中AIMS最为稳健。基因组DNA污染对鉴定Basal-like亚型的一致性影响较小(表3)。虽然Basal-like亚型鉴定的一致性较高,仍有部分样本在鉴定Basal-like亚型时不一致,而鉴定其他亚型时一致性更差5。尽管乳腺癌分子亚型的建立为乳腺癌精准医疗提供了基础,然而如何确立一套准确的鉴定分子亚型的参数与指标仍需要进一步探索。
本研究阐明了在RNA-seq数据中基因组DNA污染及不同SSP对Basal-like亚型乳腺癌鉴定的一致性与稳健性,可为医生在对患者进行临床决策时提供一定参考。
参考文献:
1. Sørlie, T. et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proceedings of the National Academy of Sciences 100, 8418 (2003).
2. Hu, Z. et al. The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genomics 7, 96 (2006).
3. Parker, J.S. et al. Supervised Risk Predictor of Breast Cancer Based on Intrinsic Subtypes. Journal of Clinical Oncology 27, 1160-1167 (2009).
4. Paquet, E.R. & Hallett, M.T. Absolute assignment of breast cancer intrinsic molecular subtype. J Natl Cancer Inst 107, 357 (2015).
5. Weigelt, B. et al. Breast cancer molecular profiling with single sample predictors: a retrospective analysis. The Lancet Oncology 11, 339-349 (2010).
6. Lusa, L. et al. Challenges in projecting clustering results across gene expression-profiling datasets. J Natl Cancer Inst 99, 1715-1723 (2007).
7. Van Peer, G., Mestdagh, P. & Vandesompele, J. Accurate RT-qPCR gene expression analysis on cell culture lysates. Scientific Reports 2, 222 (2012).
8. Naderi, A. et al. Expression microarray reproducibility is improved by optimising purification steps in RNA amplification and labelling. BMC Genomics 5, 9-9 (2004).
9. Sørlie, T. et al. The importance of gene-centring microarray data. Lancet Oncol 11, 719-720; author reply 720-711 (2010).
10. Gendoo, D.M. et al. Genefu: an R/Bioconductor package for computation of gene expression-based signatures in breast cancer. Bioinformatics 32, 1097-1099 (2016).

客服QQ:30444492琼网文【2021】1550-113号
增值电信业务经营许可证:琼B2-20210322
出版物经营许可证:新出发龙华出字第(2021)009号
广播电视节目制作经营许可证:(琼)字第00779号
版权所有 ©2002-2024 期刊网(www.qikanchina.com) 琼ICP备2021005105号



