1. 广州市市场监督管理局( 广东 广州 ) 510620 2. 诺诚健华(广州)生物科技有限公司 (广东 广州) 510000
摘要:科学开展风险评估是实施药用辅料风险管理的前提,由于辅料的特殊性和复杂性,目前缺乏具体的评估方法。本文在欧盟辅料风险评估指南的基础上,提出一种基于辅料属性参数的风险指数评分系统,用于总体评估辅料安全性、功能性和质量属性等与制剂质量相关的风险水平。该方法经过验证,具有普遍适用性,可为药品上市许可持有人有效开展辅料风险评估工作提供借鉴和参考。
关键词:药用辅料 风险评估
正文
1 背景
药用辅料涉及药物处方组成,是药物药效的基础,其成分工艺复杂多样,作用机理难以确定,一直是监管的难点。当前,世界主要国家包括我国,对药用辅料的管理都是采用以DMF形式备案以及和制剂申报关联审评审批的模式。该模式下,由药品上市许可持有人(MAH)全面负责其所用辅料的安全和质量管理。伴随着科技的进步,对药用辅料相关性质和变异性认识理解的深入,世界各国监管部门都在积极引导药品MAH建立辅料风险评估思维,以期通过基于风险的方法来保证药品制剂的质量疗效。2016年,欧盟发布了辅料风险评估指南[1],作为药品MAH对辅料进行风险评估的基本原则,要求药品MAH采用科学的评估工具对使用的辅料开展风险评估,并建立风险控制策略。这是目前为止世界范围内药品监管部门发布的关于辅料风险评估的唯一官方技术指南。
越来越多的国家开始重视药用辅料的风险评估,强调辅料的合理选用及变更分级管理的风险评估依据。近5年来,我国药用辅料的监管法规体系和技术标准体系持续与国际接轨,密集出台了《关于药包材药用辅料与药品关联审评审批有关事项的公告》、《动物来源药用辅料指导原则》、《预混与共处理药用辅料质量控制指导原则》、《药用辅料功能性指标研究指导原则》等一系列法规和技术指导文件,已具备了开展辅料风险评估的基础。当前,我国已上市品种使用的药用辅料500多种,药用标准的约200多种,其余的为化工试剂或食用产品,标准参差不齐;且90%以上的药物制剂品种上市前未对其选用的辅料适用性和安全有效性进行过研究[2]。相对而言,我国药用辅料的使用情况更为复杂,行业、监管部门关于药用辅料实施风险管理的诉求逐步提上日程。
欧盟辅料风险评估指南虽然给出了参数清单,但没有具体说明如何使用这些参数进行评估,没有具体的评价方法和实施工具。如何全面有效的排查辅料的使用风险,并依据药物制剂的质量要求(CQA)采取与辅料风险程度相对应的控制措施,特别是针对早前已审批上市的品种,是当前药品上市持有许可人实施全面药品质量管理的难点和一项重要的研究课题。本研究旨在欧盟辅料风险评估指南的基础上,建立一种通用的、适合各类辅料的数值评分系统,可用于总体评估辅料的原料控制、生产、检验、安全性、功能性、稳定性等各类与制剂质量相关的风险水平,为药品上市许可持有人开展辅料风险评估提供有效工具。
2 模型的建立
风险管理方法在制药生产中已经实施了十多年,根据国际人用药品注册技术协调会(ICH)发布的 《ICH Q9-质量风险管理》推荐的风险评估方法和工具,相关工具可以组合和拓展。本研究拟在风险系数法的基础上,采用特殊特性识别准则开发一个定量的风险评估方法。
建立药用辅料定量风险评估方法有3个步骤:①基于药用辅料需要评价的属性,确定用于计算风险系数(RPN)三个维度的坐标内容。②评分体系的开发。对欧盟风险评估指南给出的参数进行研究,设计每个参数下自变量和评分标准,以及RPN对应的风险等级。③明确风险排序准则及对应的控制策略。
2.1确定用于计算风险优先顺序数(RPN)三个维度
欧盟风险评估指南中详细列举了需要评估的可能造成辅料使用风险的因素,评估范围涵盖辅料生产源头到制剂应用环节,主要包括辅料所呈现的质量、安全和功能方面的风险,以及辅料生产商执行GMP水平的风险。辅料生产商执行GMP水平的风险可归入质量风险。因此,RPN计算的三个维度确定为:辅料安全性指标(S),辅料功能性指标(F)、辅料质量指标(Q)。
2.2 风险评分体系的开发
以广州某工厂的数据为基础,从更易理解的角度出发,采用等差序列1,2,3进行赋值,自变量参数的设计结合了具体产品的特点和功能且具有清晰的识别特征。
2.2.1辅料安全性指标(S)赋值表的开发
基于ICHQ8质量源于设计的理念,辅料安全性指标(S)是针对辅料自身的安全性项目。欧盟指南重点关注了辅料来源(动植物)、制备工艺可能带入的病毒、微生物污染及杂质污染对药物制剂的影响情况。一般情况下认为,有机无机杂质多于或等于0.10%,或其具有生理学效应,在极低水平时认为是不安全的,则列为高风险。在病毒、微生物污染的参数指标设计中,还需要进一步考虑到制剂成品的质量标准以及辅料在制剂生产的降解路径和加工条件的机理。比如大容量注射剂所用的辅料,一定程度(超过可能导致内毒素的不可控)的微生物负荷是允许的,因为在制剂制备中,最终灭菌工序是可以完全去除一定程度范围内由前期工序带入的微生物污染。再如病毒的污染对生物制品影响较大,但对非生物制品药物的影响就较小。
相关安全性评估数据主要来自供应商资料的收集、例如S1基于供应商的TSE/BSE证明性文件,S2-S6来源于供应商的工艺资料,如果供应商无法提供工艺资料,有必要对供应商(生产商)的现场进行审计工作,检查相关的信息便于判断安全性风险。相关设计参数和赋值见表1。
表1 辅料安全性指标(S)赋值表
序号 | 欧盟推荐项目 | 设计的参数指标 | 分值 |
S1 | 海绵状脑病 | 无动物源性原料用于生产辅料 | 1 |
动物源性原料用于生产辅料 | 3 | ||
S2 | 病毒污染 | 不可能 | 1 |
可能但对该剂型不太可能 | 2 | ||
可能并且对该剂型可能性极大 | 3 | ||
S3 | 微生物/内毒素/热源污染 | 辅料是合成的并对微生物有抑制作用 | 1 |
可能但对该剂型不太可能 | 2 | ||
可能并且对该剂型可能性极大,例如生产过程用到动物源性原料且该原料可促进微生物的生长。 | 3 | ||
S4 | 来自原料的杂质污染(例如黄曲霉、杀虫剂) | 无植物源性的原料用于生产辅料 | 1 |
植物源性的原料用于生产辅料 | 3 | ||
S5 | 工艺杂质污染(催化剂、有机残留) | 低风险 | 1 |
高风险 | 3 | ||
S6 | 其它工艺杂质(非专用设备设施中其他工艺带入杂质的可能性) | 低风险 | 1 |
高风险 | 3 |
2.2.2辅料功能性指标(F)赋值表的开发
欧盟认为,辅料功能性指标(F)的评估应根据剂型、药物处方组成、用药方式和辅料的作用综合评估其影响。当设计各参数指标时,应建立客观标准。由于辅料的实际应用情况复杂,本研究主要结合法规要求及常规用法划分不同风险区间。按照定义的不同,辅料可分为不同类型。依据是否有使用经验和支持性数据来划分,可简单分为常规辅料和新型辅料。常规辅料的相关使用信息(如单剂量的最大用量,有害作用水平等)可在FDA非活性成分数据库(IID),日本《医药品添加物事典》,粮农组织/世卫组织食品添加剂联合专家委员会数据库,国家药审中心辅料数据库等中查询借鉴[3],新型辅料则无任何可供参考借鉴的经验和数据,其风险程度应有不同 。对“剂型”风险划分依据的是国际药用辅料协会(IPEC)按给药途径对辅料的风险排序。“辅料与药品关键质量属性的关系”则是依据我国已上市化学药品、中药药学变更研究技术指导原则,按照微小、中等和重大变更规定项下辅料总量变更总和的范围对其风险等级进行设定。辅料功能性受制剂处方组成和比例影响较大,且影响因素复杂。因此,评估“辅料功能”、“辅料在处方中的组成比例”、“辅料每日服用量”、“辅料是否属于混合物”的风险,本研究着重强调了占比关系。目前已发布的化药、中药生产工艺研究技术指南规定了不同辅料在处方中的占比要求,且已有文献报道辅料每日最大用量情况对辅料进行风险低中高等级划分[4],但涉及众多的辅料,每种辅料的最大用量各不相同,作为通用的评估模板并不适用。故本文采用F=1(概率>80%),2(概率在10%-20%),3(概率<10%)对占比进行设定。辅料不完全是非活性成分,有些辅料具有一定的生物活性,且对制剂的安全疗效也有着重要影响, 如脂肪乳注射液中的卵磷脂,儿童用药中慎用的阿斯巴甜等,有的是正向的作用,有的是副作用,在风险划分中也应特别关注。
相关功能性评估数据主要来自企业产品研发数据。相关设计参数和赋值见表2。
表2 辅料功能性指标(F)赋值表
序号 | 欧盟推荐项目 | 设计的参数指标 | 分值 |
F1 | 类型 | 常规辅料 | 1 |
新型辅料 | 3 | ||
F2 | 剂型 | 外用(局部)制剂 | 1 |
口服、阴道和直肠用制剂、肺部、吸入用药 | 2 | ||
无菌制剂 | 3 | ||
F3 | 辅料与药品关键质量属性的关系 | 辅料变化超过10%会导致药品关键质量属性发生变化 | 1 |
辅料变化超过5%,但在10%内会导致药品关键质量属性发生变化 | 2 | ||
辅料变化未超过5%会导致药品关键质量属性发生变化 | 3 | ||
F4 | 辅料功能(按照药典章节) | 辅料特定功能在制剂中应用 | 1 |
该辅料的特定功能在制剂应用中不超过2个(≤2) | 2 | ||
该辅料的特定功能在制剂应用中超过3个(≥3) | 3 | ||
F5 | 辅料在处方中的组成比例 | X≤10% | 1 |
10%<X<40% | 2 | ||
X≥40% | 3 | ||
F6 | 辅料每日服用量 | X≤100mg | 1 |
100mg<X<500mg | 2 | ||
X≥500mg | 3 | ||
F7 | 辅料是否属于混合物 | 单一物品 | 1 |
两种物品混合物 | 2 | ||
超过两种物品的混合物 | 3 | ||
F8 | 辅料的生物活性 | 辅料与生物医学需求无直接关系 | 1 |
辅料与生物医学需求有直接关系 | 3 |
2.2.3辅料质量指标(Q)赋值表的开发
除了确定辅料安全性、功能性等固有风险的可接受水平外,辅料质量风险的持续性考察也是整体研究的其中一个重要维度,尤其是针对已上市药物。欧盟推荐的辅料质量指标(Q)主要涉及药品MAH对辅料全产业链条的质量控制和质量检测,相关参数都可以通过审计和检测的数据进行表征。收集和评估这些数据,可掌握不同辅料生产商之间或同一生产商不同批次间质量差异的风险程度。质量体系管控相关参数(Q1-Q6)主要包括辅料的生产工艺变更管理、无菌生产情况、供应链及运输管理、辅料包装完整性以及质量缺陷等内容,按Q=1(概率>80%),2(概率在10%-20%),3(概率<10%)进行赋值。质量检测相关参数(Q7-Q8)是辅料相关的物理、化学、微生物学性质,可以根据实际情况分项进行考察。以辅料功能性检测指标为例,粉末类辅料需要重点考虑物理参数(例如堆积密度,表面积,颗粒形状和粒径分布);液体辅料可能需要评价其PH和粘度指标;对于所有的聚合物辅料,则需要重点考虑分子量分布的影响;根据这些指标不同批次检测数据的波动,以有无显著性差异,有无不合格情况进行风险赋值。
相关质量指标评估数据主要来自药物制剂企业的辅料供应商日常管理质量体系,例如日常检测结果、OOS、偏差、供应商投诉等。相关设计参数和赋值见表3。
表3 辅料质量指标(Q)赋值表
序号 | 欧盟推荐项目 | 设计的参数指标 | 分值 |
Q1 | 观察到辅料供应商技术性的工艺变更 | 未有技术性工艺变更 | 1 |
出现技术性工艺变更 | 3 | ||
Q2 | 无菌辅料的无菌保障 | 辅料生产过程有采取无菌保障措施 | 1 |
无菌保障系统存在部分问题 | 2 | ||
未采取无菌保障措施 | 3 | ||
Q3 | 供应商及运输管理 | 未违反过建立的运输条件管理措施 | 1 |
违反过(1次/年)建立的运输条件管理措施 | 2 | ||
违反过(大于2次/年)建立的运输条件管理措施 | 3 | ||
Q4 | 供应链的复杂性 | 直接从厂家运输到用户 | 1 |
直接从代理商运输 | 2 | ||
通过2个或以上的中间商运输 | 3 | ||
Q5 | 包装完整性保证 | 无包装完整性问题 | 1 |
已识别极少数的包装稳定性问题 | 2 | ||
多起包装完整性问题被识别 | 3 | ||
Q6 | 质量缺陷或掺假 | 未发现质量缺陷或掺假事件 | 1 |
少量非严重质量缺陷被发现(小于10%) | 2 | ||
掺假事件被识别或部分质量缺陷被发现(大于10%) | 3 | ||
Q7 | 关键质量属性(化学性质、物理性质、微生物学性质等各类需要关注的属性) | 无关键质量属性测试出问题,不存在显著差异 | 1 |
关键质量属性在合格范围内,但不同批次间存在显著差异 | 2 | ||
出现过关键质量属性测试不合格问题 | 3 | ||
Q8 | 辅料的稳定性 | 辅料的稳定性没有问题 | 1 |
辅料的稳定性出现问题 | 3 |
2.2.4 风险耐受度的开发
计算方法:风险系数RPN=S×F×Q。
RPN的分数范围为:1-27。按照四分之一分位数计算为5.4,二分之一分位数为13.5,可确定低风险: 1 ≤ TRI(S x Q x F) < 5.4 ,中风险: 5.4 ≤ TRI <13.5,高风险: 13.5 ≤ TRI ≤ 27。
赋值:S1-S6,F1-F8,Q1-Q8每项属性打分,适用的项目打分后算平均数,保留1位小数,数据修约原则为只进不退;如果信息不全的按照高分数计算。
2.3 风险排序准则及控制策略
RPN值越高,提示风险越高。基于目前的认知,低风险视为广泛可接受的风险,无需进一步研究,中风险视为可接受,可能需要进一步研究,高风险不可接受,需进一步研究以降低风险。在整个辅料的风险评估过程中,药品上市许可持有人应采取系列风险控制策略降低初始风险水平,以获得最终风险概况。RPN值关联S、F、Q三个属性,其控制策略如下:
安全性指标的控制策略:如果安全性指标S分值较高且显示有不可控的风险,应与供应商沟通,加强工艺改进和过程质量监控,降低各类由于工艺可能引入的各类杂质和污染,主动介入和监控供应商工艺变更情况;如采取措施后仍未能达到降低风险的目的,可考虑更换供应商。
功能性指标的控制策略:一般来说,辅料的功能性风险比较难降低。如果功能性指标F分值较高且显示有不可控的风险,尤其是治疗范围窄、低溶解性、低渗透性等制剂中的辅料,可考虑进行辅料技术级别的变更,或处方调整。
质量指标的控制策略:质量属性要结合安全和功能属性综合判断,如果仅是质量属性分值较高,可采取更高频率的供应商审计;调整辅料质量标准参数或限度,增加检验项目等质量管理手段来降低其风险水平。如采取措施后仍未能达到降低风险的目的,可考虑停用相关辅料。
2.4 验证
2.4.1验证方法:依据本研究确定的风险评估方法,对广州某工厂XX片剂使用的辅料逐个展开分析(见表4)。处方使用了3种辅料,分别是硬脂酸镁、甘露醇、交联羧甲基纤维素钠。硬脂酸镁为润滑剂,处方占比约1%,纯度是关键控制指标;甘露醇为填充剂,处方占比约40%,还原糖是关键控制指标;交联羧甲基纤维素钠为崩解剂,处方占比5%,沉降体积、取代度等是关键控制指标。将辅料历史批次的检测数据作为基础信息,由培训师(2名)和受训人员(18名)分别进行判断赋分分值。
表4 广州某工厂XX片剂所用辅料的风险评估分析表(以甘露醇辅料为例)
辅料名称 | 甘露醇 | 分值 | |||||||
S | S1 | S2 | S3 | S4 | S5 | S6 | | | 1.9 |
1 | 1 | 2 | 1 | 3 | 3 | | | ||
F | F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | 2.3 |
1 | 2 | 3 | 2 | 3 | 3 | 1 | 3 | ||
Q | Q1 | Q2 | Q3 | Q4 | Q5 | Q6 | Q7 | Q8 | 1.3 |
1 | NA | 2 | 2 | 1 | 1 | 1 | 1 | ||
RPN值 | 5.7 | ||||||||
风险等级 | 中风险 | ||||||||
2.4.2验证结果:
(1)可理解性:参与培训的人员共18人。赋值比较合理的人员9人(最终风险值与培训师组一致,S、F、Q值不一致的不多于1个),达到可接受标准,50%的人员可以理解评价体系并合理赋分。
(2)分离度:不论培训师组还是受训人员组,对各个风险的RPN分值,都可以区分优先程度不同的风险,达到可接受标准(见图5)
图5 受训人员和培训师RPN平均值对比图
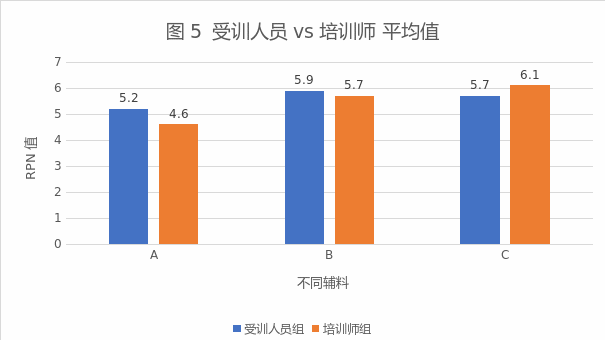
(3)准确度:根据RPN值,确定3个辅料在该剂型中的风险的排序,受训人员组和培训师组不一致的不超过1个,达到可接受标准。其中2人对其中1个辅料的赋分与培训师组不一致,定义为中风险,培训师定义为低风险。
3结论:
(1)本研究确定的风险评估方法验证结果符合可接受标准。设计的各指标参数能客观全面的评估辅料的基本风险水平,以RPN公式确定分值可以有效的区分高、中、低风险类别,并从辅料的安全性、功能性和质量属性三个方面实施风险管控,具有普遍适用性。
(2)开展持续的风险评估。在最初的风险概况确定之后,药品上市许可持有人企业应持续使用该工具开展药用辅料的风险评估,并和之前的评估情况进行差距分析,观察质量趋势,对应风险采取相应降低或减少风险的措施,实现动态风险管理。
参考文献:
[1]IPEC. Guidelines of 19 March 2015 on the formalized risk assessment for ascertaining the appropriate good manufacturing practice for excipients of medicinal products for human use. https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:52015XC0321(02)&from=RO
[2]梁毅,对加强我国药用辅料监管的思考[J],医药工程设计,2013年01期
[3]马俊威,安娜,药用辅料风险评估及管理的策略[J],中国医药工业杂志,2020,51(8):1080-1084
[4]RUBAN O, PIDPRUZHNYKOV Y,KOLISNYK T. Excipient risk assessment: possible approaches to assessing the risk associated with excipient function[J],J Pharm Invest,2018,48(4):421-429
9

客服QQ:30444492琼网文【2021】1550-113号
增值电信业务经营许可证:琼B2-20210322
出版物经营许可证:新出发龙华出字第(2021)009号
广播电视节目制作经营许可证:(琼)字第00779号
版权所有 ©2002-2024 期刊网(www.qikanchina.com) 琼ICP备2021005105号



