厦门医学院附属 第二医院胸心外科,福建 厦门 361021
【摘要】 目的 研究H2O2诱导正常肺上皮细胞损伤及激活上皮-间质转化(EMT)发生的修复机制。方法 体外培养人正常肺上皮细胞系Beas-2B,用H2O2处理Beas-2b 细胞,检测细胞克隆形成率和存活率;收集H2O2处理后少部分存活细胞进行再培养,采用Western Blot 检测EMT标志蛋白E-钙粘蛋白(E-cadherin)和波形蛋白(Vimentin)表达量,及上游PTEN-ILK信号通路关键因子的表达量。结果 H2O2显著降低了Beas-2B细胞克隆形成(p<0.05),少部分存活细胞的E-cadherin和PTEN蛋白表达量显著降低,Vimentin、ILK和Akt S473蛋白表达量显著增加。结论 H2O2可诱导 Beas-2B细胞大量损伤及死亡,同时诱导EMT发生而启动病理性修复,其机制可能是H2O2通过PTEN-ILK的信号参与EMT的激活和持续活化。
【关键词】人正常肺上皮细胞;上皮-间质转化;过氧化氢、病理性修复
放射性肺损伤(radio-pulmonary lesion)是指由于对胸部的恶性肿瘤(如肺癌、乳腺癌等)进行放射治疗所引起的并发症。肺是一个对放射线敏感的器官,其损伤程度根据放射剂量、放射部位和放射范围的不同而不同。大面积、高剂量的放疗,肺损伤的发生率和严重程度显著增大[1]。 一般认为放射性肺损伤有两种表现形式,即早期的放射性肺炎和后期的放射性纤维化。放射性肺炎主要发生于放疗后的前1-3个月;放射线诱发的纤维化是肺损伤的一个延迟反应,发生在放射后的几个月到几年,病程呈进行性加重[2]。放疗后正常肺上皮细胞病理性修复在肺纤维化发生中扮演重要角色,探索参与肺上皮细胞损伤和病理性修复的分子机制可能有助于为胸部肿瘤放疗后并发症的预防和治疗提供科学依据。
当X线照射胸部时,肺上皮损伤和修复即刻就被启动了。一般而言,创面修复是通过局部祖细胞增殖和募集骨髓干细胞进行表型分化,以重建结构和功能完整的上皮细胞层[3]。新近研究显示上皮细胞和内皮细胞可通过上皮/内皮-间质转化(epithelial/endothelial–mesenchymal transition,EMT/ EndMT)的方式参与组织损伤修复[4]。但在放射性肺损伤后修复过程中是否也激活了EMT?EMT是如何被激活?又是如何及参与放疗后肺损伤修复?这些问题还有待进一步研究。
既往有研究者提出慢性氧化应激反应可能是放射后正常肺组织损伤的重要诱因[5],如临床观察发现肺癌患者放射治疗后肺组织出现慢性氧化应激反应[6];在肺损伤动物模型中也观察到慢性氧化应激反应[7-8]。而研究证实NADPH氧化酶NOX4是慢性缺氧应激反应中唯一个表达增高的氧化-还原酶[9-10]。Hecker等在两个不同的小鼠肺损伤模型的研究中均发现NOX4依赖生成的活性氧自由基(reactive oxygen species,ROS)-过氧化氢(H2O2)是成纤维细胞分化所必需的[11],且ROS可诱导胚胎干细胞分化[12]。既往研究已表明,NADPH氧化酶NOX4表达水平在经X光照射的肺组织(包括远侧肺泡)中逐渐增加[13]。而H2O2是NOX4的唯一产物,以上研究提示H2O2可能参与放射性肺损伤及修复,但目前尚缺乏直接证据。
基于此,本研究通过体外培养肺上皮细胞Beas-2B,并采用H2O2处理后观察细胞克隆形成和存活率,对处理后少数存活细胞进行再培养,并检测EMT相关蛋白的表达水平,以探讨H2O2诱导肺上皮细胞的损伤及激活EMT发生在损伤修复中的作用。
1. 材料和方法
1.1细胞培养:从ATCC购买人正常肺上皮细胞系Beas-2B,用含有上皮细胞生长因子-支气管上皮细胞生长培养基,置于37℃,5%CO 2的培养箱中培养。
1.2克隆形成实验: 将种板于10厘米培养皿的Beas-2B细胞中用1mM H2O2孵育30分钟。重悬并制成5×104单细胞悬液与基质胶以1:1混合,接种在6cm的培养皿中,培养箱中温育15天后,将细胞用的碘硝基四唑紫溶液(1mg/ml)孵育过夜。在显微镜下计数含50个细胞以上的细胞集落。计算克隆形成率(%)和细胞克隆存活率:克隆形成率=(克隆数/接种细胞数)100%;细胞克隆存活率=处理组克隆形成率/对照组克隆形成率。每个实验设3个重复,实验重复3次。
1.3 Western-blot检测:首先将Beas-2B细胞在含有1mM H2O2培基中孵育3天,然后换正常培基再培养1周后收集细胞,提取总蛋白。参照Zhang等[13]进行蛋白免疫印记实验。一抗E-cadherin、Vimentin、ILK1、PTEN、磷酸化Akt S473为CST公司兔抗鼠多克隆抗体,内参照α-tubulin一抗为Abcam公司的兔抗鼠单克隆抗体。用辣根过氧化物酶标记的羊抗IgG 为二抗。
1.4 统计分析 采用SPSS17.0统计软件包对数据进行统计分析,计量数据以均值±标准差(x±SD)表示,组间比较采用独立样本 t 检验,设p<0.05为差异有统计学意义。
2. 结果
2.1 H2O2处理Beas-2B细胞系后细胞的生长增值情况
克隆形成试验结果显示,1mM H2O2处理30min后显著降低了Beas-2B细胞的克隆形成(t=19.43,p<0.001)(图3);但结果仍有少部分细胞存活下来了,克隆存活率为14.35%。
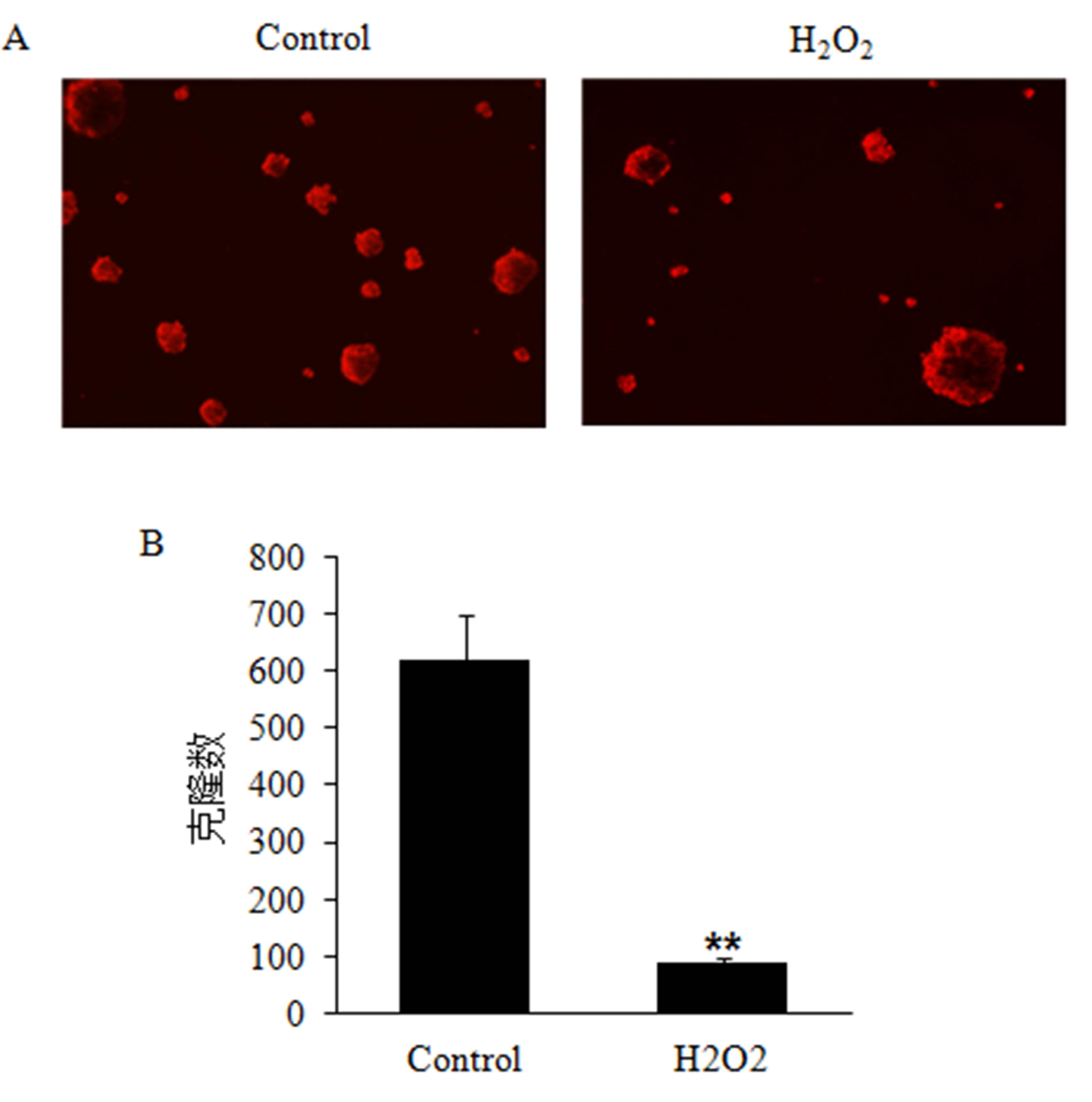
图1 H2O2处理后Beas-2B细胞的克隆形成试验
A:荧光显微镜下对照组和H2O2处理组Beas-2B的克隆形成;B:对照组和H2O2处理组Beas-2B的克隆数比较。**与对照组比较,P<0.01。
2.3 H2O2处理Beas-2B后存活细胞EMT相关分子表达水平的变化
Western Blot结果显示,与对照组比较,H2O2处理后再培养细胞的E-cadherin蛋白表达量显著降低了(t=16.99,p<0.001),而vimentin蛋白表达量显著增高了(t=-14.33,p<0.001)(图 4A);结果还显示,与对照组比较,与EMT激活相关蛋白PTEN在存活下来的Beas-2B细胞中显著降低了(t=8.56,p<0.001),而ILK(t=-8.97,p<0.001)和Akt S473(t=-3.81,p=0.003)蛋白显著增加了(图 4B)。
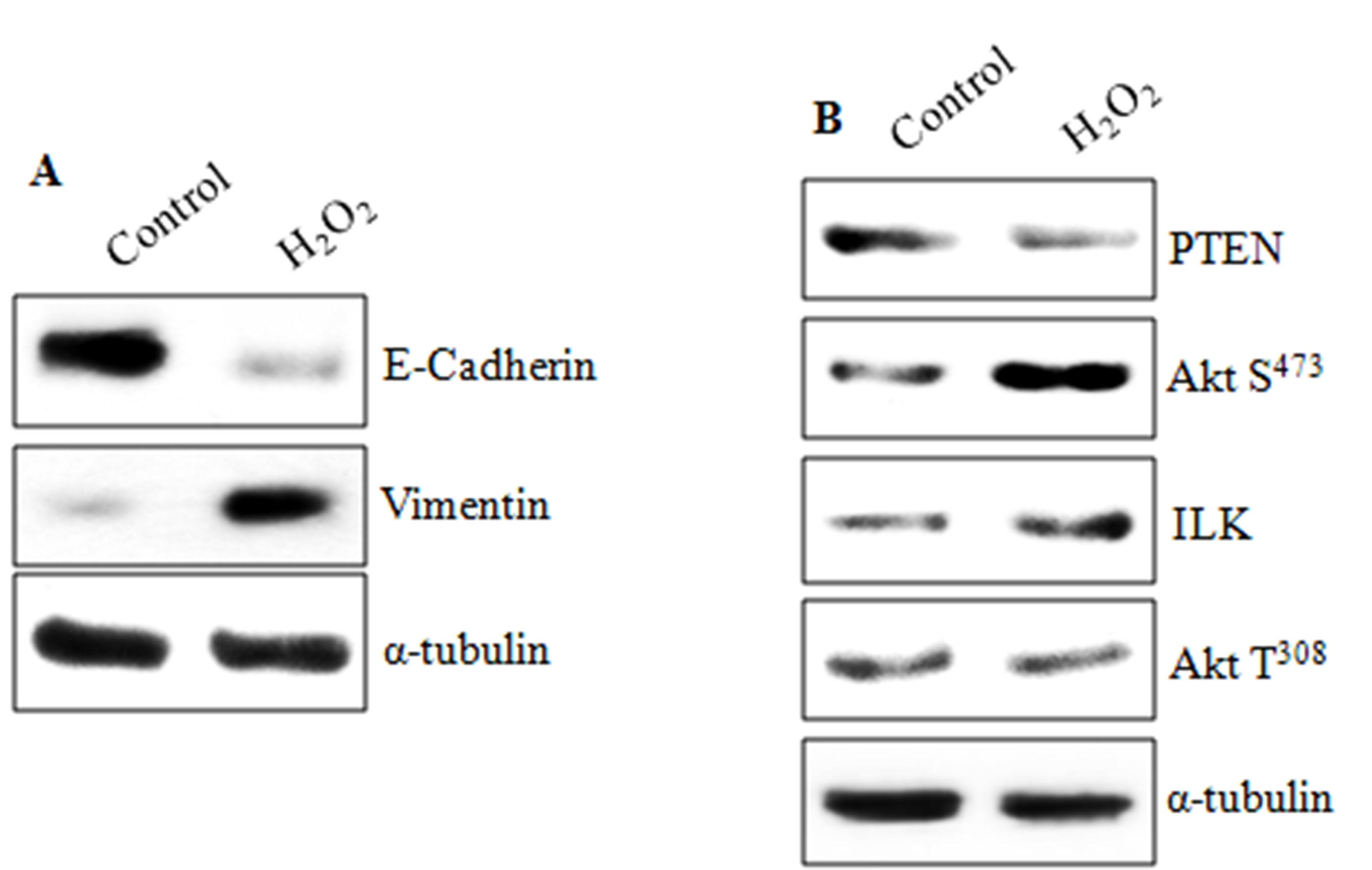
图 4 H2O2诱导的EMT 时Beas-2B内相关分子表达水平
A:H2O2处理组和对照组(Control)Beas-2B细胞内E-cadherin和vimentin蛋白的Western Blot条带图 B: H2O2处理组和对照组Beas-2B细胞内PTEN、Akt S473、ILK、Akt T308蛋白的Western Blot条带图;α-tubulin作为内参照。
3. 讨论
本研究首先采用过氧化氢(H2O2)处理人正常肺上皮细胞Beas-2B细胞,克隆形成试验结果显示,1mM H2O2处理30min后显著降低了Beas-2B细胞的克隆形成;但结果仍有少部分细胞存活下来了,细胞克隆存活率为14.35%。结果提示H2O2诱导了人正常肺上皮细胞大量死亡。既往研究已表明,NADPH氧化酶NOX4是细胞慢性缺氧应激反应中唯一个表达增高的氧化-还原酶[9-10],且NOX4表达水平在经X光照射的肺组织(包括远侧肺泡)中逐渐增加[13]。而H2O2是NOX4的唯一产物,提示NOX4产生的H2O2可能是放射性肺损伤过程中对周围正常肺组织细胞造成损伤的关键因子。
本研究在克隆形成试验时发现,1mM H2O2处理30min后仍有少部分Beas-2B细胞存活下来了,为研究这些存活下来细胞特性,本研究首先将Beas-2B细胞在含有1mM H2O2培基中孵育3天,然后换正常培基再培养1周后收集细胞,Western Blot结果显示H2O2处理后再培养细胞的E-cadherin蛋白显著降低了,而vimentin蛋白表达量显著增加了,而E-cadherin蛋白的降低和vimentin蛋白的增加被认为是EMT激活的典型特征[14]。提示1 mM H2O2处理后存活下来的Beas-2B细胞具有EMT细胞的特征。Western Blot结果还显示,与EMT激活相关蛋白PTEN在存活下来的Beas-2B细胞中显著降低了,而ILK和Akt S473蛋白显著增加了。以上结果提示H2O2可能通过PTEN-ILK信号通路调节EMT发生。
EMT发生可分为3种类型:I型EMT,主要发生在个体生命早期,参与胚胎形成与组织器官的发育;II型EMT,通常发生在组织受到创伤后产生成纤维细胞及其他相关细胞进行组织重建与伤口愈合;III型EMT,主要与肿瘤的侵袭性和转移相关[15-16]。Ⅰ型主要出现在生理情况下,Ⅱ型和Ⅲ型主要在病理情况下出现。而器官(心、肝、肺、肾)纤维化主要与II型EMT发生密切相关[15]。既往研究认为炎症反应是肺纤维化的重要诱因,持续的验证反应能导致纤维化,但临床上抗炎治疗却不能明显改善肺纤维化[17]。提示炎症可能不是导致纤维化的必要条件。Willis认为肺纤维化不是炎症的持续过程,而是病理性的损伤修复过程,肺上皮细胞的损伤修复在肺纤维化过程中起到核心作用[18]。在此研究中,我们证明了:(1)人正常肺上皮细胞Beas-2B细胞可以在经过高浓度H2O2处理后仍有部分细胞存活下来。这些存活下来的上皮细胞E-cadherin的表达被抑制,而vimentin蛋白高表达,这些结果为H2O2激活损伤后EMT的修复机制提供了直接的证据。为明确H2O2激活EMT的具体分子机制,本研究检测了存活细胞中与EMT相关的因子。既往研究显示,X射线照射后,肺组织内PTEN表达增加, 而ILK的表达和/或Akt磷酸水平随后开始降低,这些因子都是参与放射诱导的上皮细胞和内皮细胞凋亡的关键因子
[13]。与以往研究相反,本研究发现经H2O2处理Beas-2B从而诱发EMT过程中,PTEN蛋白的表达水平显著降低了,而ILK蛋白表达及Akt的473位丝氨酸磷酸化水平显著增加了。但本研究结果与既往研究一致表明H2O2在细胞信号传导中起重要作用[19,20],诱导的EMT参与了细胞损伤后修复。
综上所述,本研究结果表明过氧化氢可诱导肺上皮细胞损伤,同时通过PTEN-ILK/Akt信号通路激活并持续活化EMT,从而参与细胞损伤后病理性修复。
参考文献
[1] Ding N H, Li J J, Sun L Q. Molecular Mechanisms and Treatment of Radiation-Induced Lung Fibrosis[J]. Current Drug Targets, 2013, 14(11):1347-56.
[2] Brush J, Lipnick S L, Phillips T, et al. Molecular mechanisms of late normal tissue injury[C]. Seminars in Radiation Oncology. PubMed, 2007:121-30.
[3] Crosby L M, Waters C M. Epithelial repair mechanisms in the lung[J]. Ajp Lung Cellular & Molecular Physiology, 2010, 298(6):715-31.
[4] Wynn T A. Cellular and molecular mechanisms of fibrosis[J]. Journal of Pathology, 2008, 214(214):199-210.
[5] Zhao W, Robbins M E. Inflammation and chronic oxidative stress in radiation-induced late normal tissue injury: therapeutic implications[J]. Current Medicinal Chemistry, 2009, 16(2):130-43.
[6] Beinert T, Binder D, Stuschke M, et al. Oxidant-induced lung injury in anticancer therapy[J]. European Journal of Medical Research, 1999, 4(2):43-53.
[7] Fleckenstein, Gauter-Fleckenstein K ;, Jackson B ;, et al. Using biological markers to predict risk of radiation injury[C]. Seminars in Radiation Oncology. 2007.
[8] Anscher M S, Thrasher B, Rabbani Z, et al. Antitransforming growth factor-beta antibody 1D11 ameliorates normal tissue damage caused by high-dose radiation[J]. International Journal of Radiation Oncology Biology Physics, 2006, 65(3):876-81.
[9] Cheng G, Cao Z, Xu X, et al. Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5[J]. Gene, 2001, 269(1-2):131-40.
[10] Mittal M, Roth M P, Hofmann S, et al. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature[J]. Circulation Research, 2007, 101(3):258-67.
[11] Hecker L, Vittal R, Jones T, et al. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury[J]. Nature Medicine, 2009, 15(9):1077-81.
[12] Barcelloshoff M H, Dix T A. Redox-mediated activation of latent transforming growth factor-beta 1[J]. Molecular Endocrinology, 1996, 10(9):1077-83.
[13] Yu Z, Xiuwu Z, Rabbani Z N, et al. Oxidative stress mediates radiation lung injury by inducing apoptosis [J]. International Journal of Radiation Oncology Biology Physics, 2012, 83(83):740-8.
[14] Kendall RT, Feghali-Bostwick CA. Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol, 2014,5:123.
[15] Yang AH, Chen JY, Lin JK. Myofibroblastic conversion of mesothelial cells[J]. Kidney Int, 2003,63(4):1530-9
[16] Li Y, Wang J, Asahina K. Mesothelial cells give rise to hepatic stellate cells and myofibroblasts via mesothelial-mesenchymal transition in liver injury[J]. Proc Natl Acad Sci U S A, 2013,110(6):2324-9
[17] Bucala R, Spiegel LA, Chesney J, et al. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair[J]. Mol Med, 1994,1(1):71-81
[18] Quan TE, Cowper S, Wu SP, et al. Circulating fibrocytes: collagen-secreting cells of the peripheral blood[J].. Int J Biochem Cell Biol, 2004,36(4):598-606
[19] Winterbourn C C. The biological chemistry of hydrogen peroxide[J]. Methods in Enzymology, 2013, 528C(528C):3-25.
[20] Forman H J, Maiorino M, Ursini F. Signaling functions of reactive oxygen species[J]. Biochemistry, 2010, 49(5):835-42.

客服QQ:30444492琼网文【2021】1550-113号
增值电信业务经营许可证:琼B2-20210322
出版物经营许可证:新出发龙华出字第(2021)009号
广播电视节目制作经营许可证:(琼)字第00779号
版权所有 ©2002-2024 期刊网(www.qikanchina.com) 琼ICP备2021005105号



